Июл
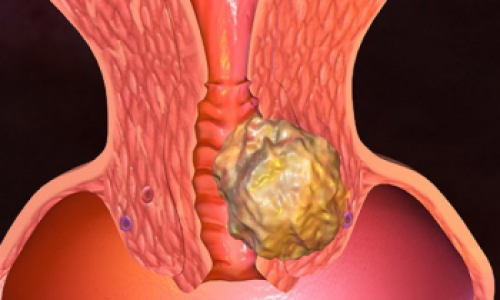
Амилоидоз
AL-амилоидоз (immunoglobulin light chains derived) — первичный амилоидоз, вызванный появлением в плазме крови и отложением в самых разных тканях организма аномальных лёгких цепей иммуноглобулинов, синтезируемых малигнизированными плазмоцитами. Тот же процесс идёт и при миеломной болезни (болезнь Рустицкого-Калера, плазмоцитома), но здесь выступает на первый план патологическая пролиферация малигнизированного клона плазмоцитов, инфильтрирующих ткани (чаще всего плоские кости и позвонки, с развитием патологических переломов). AA-амилоидоз (acquired) — вторичный амилоидоз, вызванный гиперсекрецией печенью белка острой фазы альфа-глобулина в ответ на любое хроническое воспаление. Этот процесс развивается при ревматоидном артрите, анкилозирующем спондилоартрите, бронхоэктатической болезни, хроническом остеомиелите, туберкулёзе, лепре и ряде других заболеваний. Подвидом данного амилоидоза считается ASC-амилоидоз (systemic cardiovascular), развивающийся у лиц свыше 70 лет.
Патогенез в деталях неизвестен. AF-амилоидоз (средиземноморская перемежающая лихорадка) — наследственная форма амилоидоза, с аутосомно-рецессивным механизмом передачи. Данным видом амилоидоза страдают люди, принадлежащие к определенным этническим группам, живущим по побережью Средиземного моря (евреи-сефарды, греки, арабы, армяне). Существуют разновидности амилоидоза, характерные для определенной географической местности: «португальский амилоидоз» (с преимущественным поражением нервов нижних конечностей), «американский амилоидоз» (с преимущественным поражением нервов верхних конечностей), семейный нефропатический амилоидоз, или «английский амилоидоз», протекающий с симптомами крапивницы, глухоты и лихорадки. AH-амилоидоз (hemodialisis-related) — наблюдается исключительно у больных, находящихся на гемодиализном лечении. Патогенез связан с тем, что бета-2-микроглобулин MHC I класса, в норме утилизирующийся почками, не фильтруется в гемодиализаторе и накапливается в организме. AE-амилоидоз — форма местного амилоидоза, развивающаяся в некоторых опухолях, например, в медуллярном раке C-клеток щитовидной железы. В этом случае предшественником амилоида являются патологические фрагменты кальцитонина. AJAPP-амилоидоз — островков Лангерганса при II типе сахарного диабета и инсулиноме. Амилоидоз финского типа — редкий тип амилоидоза, вызываемый мутацией гена GSN, кодирующего белок джелсолин.
Природа и механизмы развития амилоидоза до конца не ясны. Считают, что в основе развития заболевания лежит продукция белка — предшественника амилоида. Это происходит под влиянием специфического амилоид- высвобождающего фактора, выработка которого осуществляется клетками крови вследствие генетического дефекта или под воздействием провоцирующих агентов. Накопление в крови белка-предшественника совместно с нарушениями его катаболизма и (или) рассасывания приводит к выработке амилоидного белка, из которого на 96-98% состоит амилоид. Из-за недостатка знаний о факторах, влияющих на развитие системного амилоидоза, и механизма его развития болезнь принято разделять на следующие формы: первичный амилоидоз, по-видимому, представляющий собой врожденную ферментопатию, наследуемую аутосомно-доминантным путем (вероятно, имеется несколько форм врожденного амилоидоза); вторичный, возникающий на фоне длительных хронических заболеваний, характеризующихся распадом тканей и всасыванием продуктов распада (при туберкулезе легких и других органов, бронхоэктатической заболевания, хроническом остеомиелите и других заболеваниях) или значительными иммунопатологическими нарушениями (неспецифический язвенный колит, сывороточная болезнь и т. д.); идиопатический; старческий амилоидоз. Поражение пищеварительной системы встречается при любой форме амилоидоза, особенно в некоторых ее отделах (языка — макроглоссия, кишечника, печени).
По частоте отложения амилоида органы и системы можно расположить в следующем порядке: селезенка, почки, надпочечники, печень, желудочно-кишечный тракт, лимфатические узлы, поджелудочная железа, предстательная железа, сердце, головной мозг, кожа. Клинические симптомы амилоидоза зависят от преимущественной локализации амилоидных отложений и степени нарушения функции пораженного органа. Амилоидоз сердца чаще бывает первичным. Он характеризуется симптомами прогрессирующей сердечной недостаточности, параллельно с которыми (или чуть раньше) наблюдаются всевозможные нарушения проводимости и ритма сердца. Амилоидоз желудочно-кишечного тракта традиционно проявляется диарейным синдромом, реже развивается синдром мальабсорбции. При поражении пищевода характерна дисфагия. В целом нарушения всасывания приводят к развитию полигиповитаминоза и похудания вплоть до кахексии. Не не часто выявляется увеличение размеров языка, что приводит к мысли о первичном амилоидозе. Типично увеличение размеров печени и селезенки, не сопровождающееся у трети заболевших нарушениями их функций. Изолированный амилоидоз печени наблюдается крайне редко; традиционно встречаются сочетанные поражения, особо при вторичном амилоидозе. В клинике превалирует увеличение печени, которое, впрочем, может развиться на поздних стадиях заболевания. Печень при прощупывании плотная, малоболезненная. Желтуха наблюдается не часто и носит неинтенсивный характер.
Возможна внутрипеченочная обструктивная желтуха на фоне первичного амилоидоза печени. В некоторых случаях может развиться геморрагический синдром. Амилоидоз почек — поражение при вторичном амилоидозе, имеющее наиболее неблагоприятный прогноз. Клинически он подразделяется на 3 стадии: протеинурическую, нефротическую и азотемическую. Клиническая симптоматика протеинурической стадии вторичного амилоидоза малоспецифична и во многом зависит от основного заболевания (туберкулез, сифилис, бронхоэктатическая болезнь и т. д.), также от отложения амилоида в других органах. Протеинурическая стадия может продолжаться от 6 месяцев до 5 лет. Она имеет более длительное течение, если основной процесс имеет гнойный характер, а при туберкулезе, наоборот, укорачивается. Изменения выявляются только при тщательном исследовании мочевого осадка. В нефротической стадии симптомы основного заболевания отходят на второй план. Характерна триада симптомов — протеинурия, отеки, гипо- и диспротеинемия, гиперлипидемия. Развиваются олигурия и никтурия. Распространены отеки. Артериальное давление снижено, что обусловлено поражением надпочечников. Очевидно, в связи с этим снижается фильтрационная функция почек. Вследствие присоединения липоидного нефроза повышается реабсорбционная функция.
Белково-углеводная функция печени нарушена. Развиваются желудочно-кишечные нарушения. Селезенка плотная. Азотемическая стадия вторичного амилоидоза почек характеризуется развитием хронической почечной недостаточности в следствии сморщивания клубочков (амилоидно-сморщенная почка). Толчком к развитию уремии могут явиться перекрестные заболевания, травмы, тромбоз почечных сосудов и др. Снижаются мочеобразовательная, мочевыделительная функции почек. Наблюдаются полиурия, гипоизостенурия. Остаточный азот не превышает 100 мг/%. Артериальное давление в пределах нормы или некординально повышено. Отеки остаются только на ногах и лодыжках. Уровень холестерина снижается до нормы. Усиливается анемия. В этой стадии заболевания может наступить смерть заболевшего вследствие азотемической уремии или других осложнений (гнойно-септический процесс, пневмония, гнойный перитонит, тромбоз почечных вен, рожистое воспаление, абсцесс легкого и т. д.). Амилоидоз половых органов бывает как первичным, так и вторичным. Первичный амилоидоз чаще характеризуется нарушенной функцией половых желез (импотенцией у мужчин и нарушениями менструального цикла у женщин). Симптомы появляются на 2-5-м году заболевания в возрасте 20-45 лет. Продукция половых гормонов и образование сперматозоидов не нарушаются. Значительно реже выявляются поражения мочевого пузыря, матки, предстательной железы. При этом отмечаются кровотечения (маточные, макрогематурия), олигоспермия, затрудненное мочеиспускание. Симптомы амилоидоза матки и мочевого пузыря сходны с симптомами при опухолевом поражении этих органов.
Амилоидоз кожи встречается при системном амилоидозе. При этом на фоне бледной («фарфоровой») кожи (чаще лица) отмечаются желтоватые узелки или бляшки (традиционно на коже конечностей), лихенизация, реже — узлы и бородавчатые разрастания в районах кожных складок, возможны мелкие кровоизлияния. Первичный локализованный амилоидоз кожи характеризуется присутствием множественных мелких зудящих узелков на разгибательных поверхностях конечностей с лихенизацией кожи в области расчесов (амилоидный лихен) или округлыми розовато-коричневыми пятнами с непостоянным зудом, реже — опухолевидными образованиями в коже (на лице, груди, в области гениталий). Собственно кожа истончается, легко собирается в складки над этими образованиями. Встречается также вторичный локализованный амилоидоз кожи на фоне предшествующих дерматозов. Генетический (наследственный, семейный) амилоидоз наиболее распространен в государствах Средиземноморского бассейна. У нас в стране наблюдаются единичные случаи заболевания. Генетический амилоидоз начинается в первые два-три десятилетия жизни заболевшего. В клинической картине выделяют 3 основных синдрома: нефропатический, протекающий с поражением почек; нейропатический, характеризующийся преимущественным поражением нервной системы; кардиопатический, проявляющийся изменениями со стороны сердца. Клиническая картина генетического амилоидоза характеризуется лихорадкой, отеками, протеинурией, анемией, суставными и кожными изменениями, миалгиями, поражением желудочно-кишечного тракта (боли, расстройства пищеварения). Мочевой синдром может предшествовать перечисленным проявлениям заболевания или, наоборот, появляться значительно позже. У заболевших выявляются нарушения белкового состава крови, ускорение СОЭ.
Кровяное давление традиционно нормальное и не не часто не повышается даже в стадии хронической почечной недостаточности. Изменения глазного дна отсутствуют или выражены слабо. Отмечается относительно раннее снижение почечного кровотока и фильтрации. У многих заболевших отмечается увеличение печени и селезенки за счет отложения амилоидного вещества в этих органах. Функции печени нарушены. Старческий амилоидоз характеризуется развитием в преклонном возрасте симптомов сахарного диабета на фоне диабета. Это сочетается с быстро прогрессирующим атеросклерозом. При этом в бляшках на стенках сосуда при исследовании выявляется амилоид. Прогноз зависит от локализации и выраженности амилоидоза, а при вторичной форме — и от течения основного заболевания. Трудоспособность заболевших с амилоидозом почек сберегается только на протеинурической стадии. Потом они нуждаются в медико-социальной экспертизе и установлении группы инвалидности. Хроническая почечная недостаточность при амилоидозе быстро прогрессирует и приводит к смерти заболевшего. То же относится и к амилоидозу желудочно-кишечного тракта с синдромом мальабсорбции.
Диагностика первичного амилоидоза сложна и требует использования всех возможных методов. Вторичный амилоидоз органов распознается несколько легче, поскольку известно заболевание, способное привести к его развитию. Специфических лабораторных изменений при амилоидозе нет. Однако для заболевших характерно присутствие значительно повышенной СОЭ (50-70 мм/ч) и анемии (в ряде случаев отмечаются тромбоцитоз, гиперфибриногенемия). Применяют также специальные пробы на амилоид (с конгорот, метиленовой синью, которые в норме изменяют окраску мочи, тогда как у заболевших они фиксируются амилоидом и выводятся с мочой в минимальных количествах), и электрофоретическое исследование белков мочи. При амилоидозе сердца на ЭКГ регистрируется низкий вольтаж комплекса QRS. При проведении эхокардиографии отмечается сочетание повышенной эхогенности (проницаемости) миокарда и утолщения стенок предсердий, что в 60-90% случаев указывает на амилоидоз. В протеинурической стадии амилоидоза почек выявляется некординальное численность белка в моче и периодическое появление эритроцитов, которое иногда обнаруживается лишь при исследовании осадка мочи по методу Каковского-Аддиса. Кроме того, в моче определяются единичные цилиндры (гиалиновые, зернистые, восковидные) и лейкоциты. В ряде случаев некординально снижаются почечный кровоток и фильтрация. В нефротической стадии нарастает численность альбумина, среди белков мочи преобладают глобулины, особо их крупные частицы. Отмечается высокое содержание гаммагликопротеидов и альфа-липопротеидов в моче.
В осадке мочи обнаруживают небольшое численность эритроцитов, цилиндров и не не часто лейкоцитов. Наблюдается снижение уровня белков крови с нарушением их соотношения (альбуминоглобулиновый коэффициент сокращается до 1,0 и ниже), среди глобулинов преобладают альфа-2 и гамма-фракции. Может наблюдаться повышенный уровень холестерина крови. Иногда в крови обнаруживается умеренное увеличение содержания креатинина, мочевины и остаточного азота. Могут наблюдаться небольшие изменения глазного дна. Большое значение в диагностике принадлежит биопсии почек. Кроме того, уточнить диагноз помогают результаты рентгенологических и радиоиндикационных методов исследования. При амилоидозе печени лабораторное исследование выявляет характерные белковые сдвиги, особо альфа-2-глобулинемию, иммунные нарушения, часто повышена активность щелочной фосфатазы сыворотки, другие функциональные пробы мало изменены. Наибольшее диагностическое значение имеет пункционная биопсия печени.
Лечение первичного амилоидоза сопряжено с большими трудностями. На успех в лечении вторичного амилоидоза можно рассчитывать лишь в том случае, если в органах отсутствуют грубые изменения, также если устранить вызвавшие его развитие причины. Лечение включает диету, щадящий режим, по вероятности — радикальное устранение основного заболевания (остеомиелит, опухоль), назначение патогенетических и симптоматических средств. К патогенетической терапии относят назначение хингамина по 0,25 г в день, 5%-ного унитиола по 5-10 мл внутримышечно курсами по 20-30 инъекций, колхицина (эффективен при наследственных формах) по 2 мг в день, иммунодепрессантов при амилоидозе, обусловленном миеломной болезнью, и первичных формах заболевания. Симптоматические средства используются по показаниям (гипотензивные, диуретические, десенсибилизирующие средства, продукты железа, витамины группы В, сердечные гликозиды, переливание цельной крови и ее компонентов, гепатопротекторы и т. д.). При развитии хронической почечной недостаточности должен решаться вопрос о гемодиализе и трансплантации почки.